
[updated NOV 2023]
“One approach to satisfy two sets of rules” for medical device software
As stated in the last blog post, there are two sets of rules for medical device software—twice the rules, twice the confusion. My recommendation is to base your software development procedures on the international IEC 62304 standard, which is easier to understand, and then include any additional adjustments needed to meet all the FDA requirements. Here I will go into more detail about exactly what that entails and how best to ensure your SW development project checks all the boxes.
IEC 62304 Medical Device Software Standard
The IEC 62304 medical device software standard (“Medical device software—Software life cycle processes”) is comprised of five processes in five chapters (5-9):
- 5 – Software Development Process = this is the main process that SW groups are focused on and includes all the key aspects of development from planning and requirements to testing and release
- 6 – Software Maintenance Process = this is an abridged form of the main software development process and is intended to quickly release patches for SW bugs and security risks
- 7 – Software Risk Management Process = risk assessment of SW failures as well as management of SW safety features which serve as risk controls for HW failures, for use error, and for other system failures
- 8 – Software Configuration Management Process = source code control system/ development environment and how to manage builds and releases
- 9 – Software Problem Resolution Process = bug tracking system and how your organization evaluates bugs and related activities to resolve issues
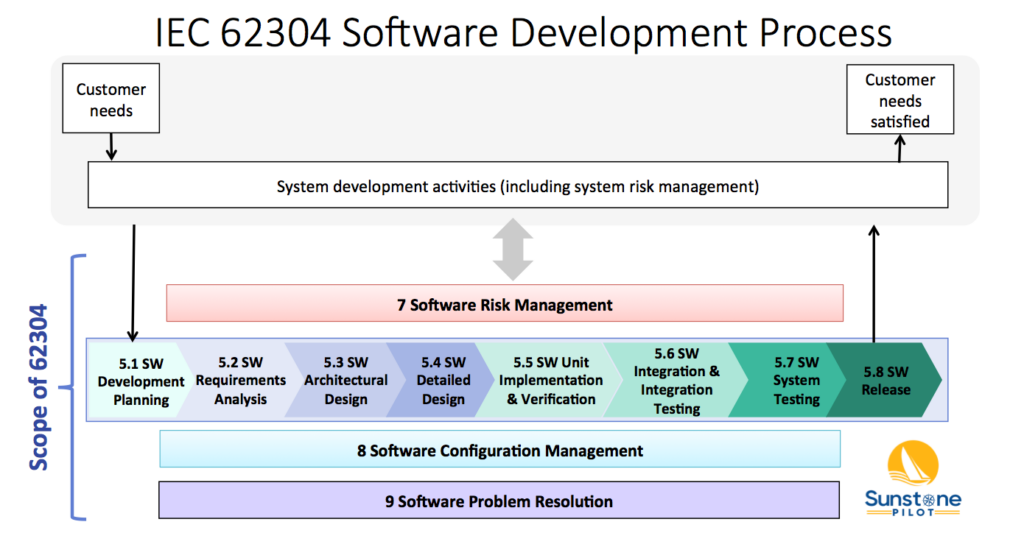
The diagram below shows 4 of these 5 processes (numbered 5-9, but missing 6) and their relationship to overall system validation. Note that the 62304 standard does not cover system validation or other system development activities—it only covers up to “SW System Testing.” FDA SW Guidances have a much broader scope, including system validation and development of non-product software.
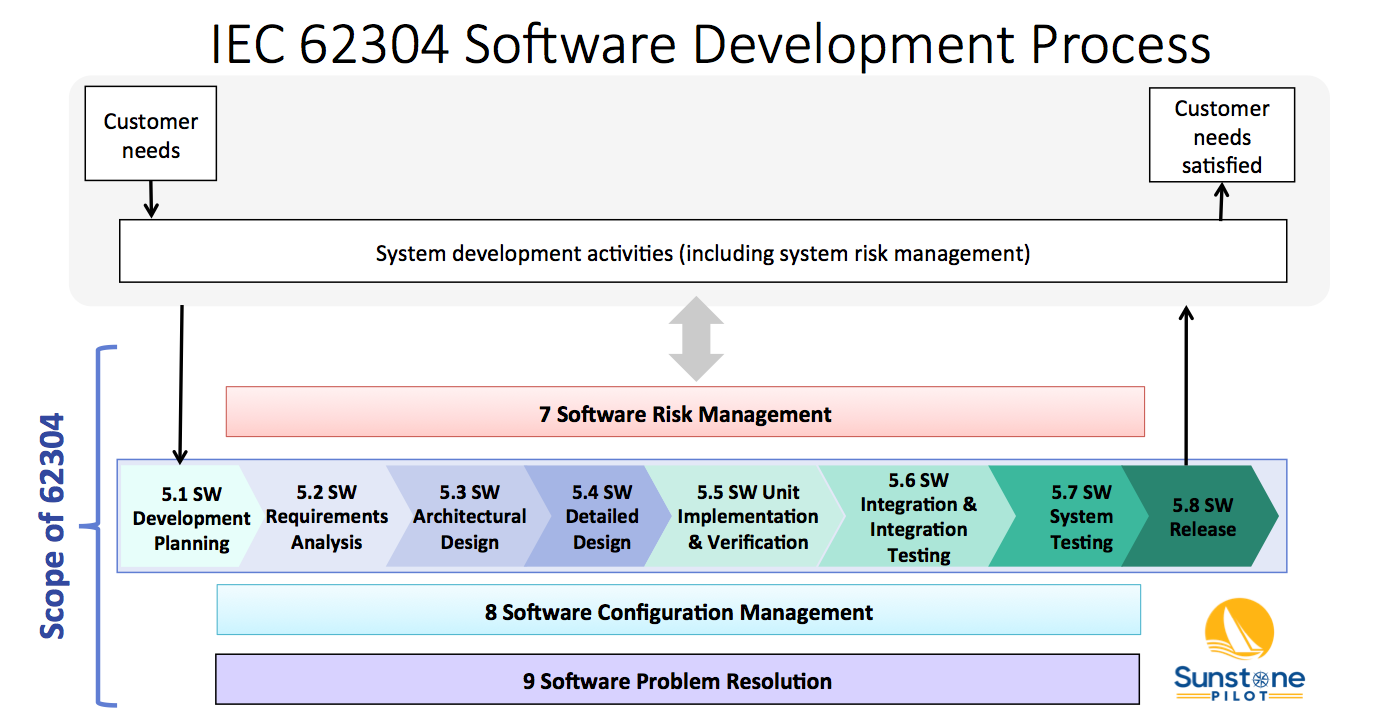
The Software Development Process (#5) consists of 8 key activities:
- SW Development Planning – defining the scope of the SW development project
- SW Requirements Analysis – decomposing system/product requirements into software requirements within the context of the system architecture design (identifying the system functions that are to be implemented in software)
- SW Architectural Design – high level design of the software, including partitioning/segregation of high-risk software, as appropriate
- SW Detailed Design – defining the SW units and their interfaces, including state diagrams, data structures, and risk controls (the software engineer’s view of how the SW will function)
- SW Unit Implementation & Verification – the core of SW development; coding and testing
- SW Integration & Integration Testing – merge software units and demonstrate that multiple software components work properly together and with the system hardware
- SW System Testing –verification of software against the SW requirements in a system environment
- SW Release – deploying a controlled, verified version of SW
These activities are described in the 62304 standard and in this diagram as a linear sequence, however, there is nothing in the standard that prevents SW teams from utilizing iterative (agile) approaches for SW development. See the AAMI TIR45 guidance for more information on using agile methods for medical device software.
Note that processes 7, 8 and 9 in the diagram above occur concurrently. The intent of the 62304 standard is that the SW team sets up configuration management and bug tracking systems at the beginning of the project and are performing risk management continually throughout development.
How these requirements of the 62304 standard are translated into specific procedures can vary considerably from company to company depending on the complexity of the software and the safety risks associated with it. For example, the appropriate methods to manage development of a small amount of firmware in a simple, low risk device would be quite different than the methods to manage software in a complex, safety-critical system.
One of the challenges in understanding the 62304 standard is that it has its own language. For example, here are some terms from it that often confuse newcomers:
- Anomaly = software bug
- SOUP = Software Of Unknown Provenance; basically Off-the-Shelf (OTS) software but can also include legacy software that was not documented
- Software Item = a software component or module (a part of a complete “software system”)
- Software Unit = the smallest software item
- Configuration Item = a software item that can be identified and tracked in the build system (e.g., a SW library from a 3rd party that is tracked by its own version number)
- Problem Resolution = bug tracking and evaluating bugs and documenting the resolution of bugs
Now that we’ve pulled the curtain back on the 62304 standard, let’s dive into software regulation from the viewpoint of FDA Guidances.
FDA Software Guidances
The FDA issued its first Software Guidance over 25 years ago, responding to issues and problems with software-controlled medical devices. The reasoning was to clearly explain FDA expectations around software development and documentation for medical device manufacturers. Over the years, they’ve updated and added more Guidances as other issues arose.
This list of Guidance documents is current as of NOV 2023 but be sure to check the FDA CDRH website for any newly released or draft guidances. Note that the term “guidance” here does not mean optional—the FDA expects you to follow their guidances exactly or have solid rationale for deviating from them. And, of course, the general FDA regulations for design controls (21 CFR 820.30) apply to all medical device (product) software. These FDA Guidances describe how to interpret those regulations for different aspects of software.
- General Principles of Software Validation; Final Guidance for Industry and FDA Staff (2002)
- Content of Premarket Submissions for Device Software Functions (2023)
- Off-The-Shelf Software Use in Medical Devices (2019)
- Cybersecurity For Networked Medical Devices Containing Off-The Shelf (OTS) Software (2005)
- Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions (2023)
- Policy for Device Software Functions and Mobile Medical Applications (2019)
- Medical Device Data Systems (MDDS): Guidance for Industry and FDA Staff (2015)
- Postmarket Management of Cybersecurity in Medical Devices (2016)
- Software as a Medical Device (SaMD): Clinical Evaluation (2017)
The first 3 are what I consider the ‘main’ software guidances (originally the oldest but two of them have been recently updated); the remaining guidances have been written to clarify more recent issues with medical device software like mobile apps, networked devices, and cybersecurity.
General Principles of Software Validation (GPSV)
This guidance, published over 20 years ago, is a general discussion of good practices for software development. It does not provide specific instructions on what must be done and since it covers a very broad scope, beyond product software, it can be difficult to translate it into specific quality system procedures. Some parts of this guidance appear to be written for an audience that is unfamiliar with software (explaining for example how software is different from hardware and describing standard practices for software testing). However, there are some particular points that stand out in the guidance:
- One of the most important points: “software testing by itself is not sufficient to establish confidence that the software is fit for its intended use.” In other words, detailed software test reports alone are not sufficient to claim that medical device software is safe and effective.
- “Because of its complexity, the development process for software should be even more tightly controlled than for hardware, in order to prevent problems that cannot be easily detected later in the development process.” Translation: you will need additional procedures and documentation for software development
- The importance of risk analysis throughout development and particular practices for safety-critical software, such as defining risk controls in the software requirements
Note that Section 6 of the GPSV guidance (Validation of Automated Process Equipment and Quality System Software) does not apply to medical device software. For validation of these types of non-product software, I recommend instead following the new draft FDA guidance Computer Software Assurance for Production and Quality System Software (2022) and the ISO/TR 80002-2:2017 guidance (Medical device software – Part 2: Validation of software for medical device quality systems). The ISO/TR 80002-2 guidance builds on the older AAMI TIR36 guidance with updates for internationalization.
Content of Premarket Submissions (UPDATED)
This guidance is important for understanding the required software documentation for regulatory submissions to the FDA. Up until 2023, the documentation depended on the software “Level of Concern” (Minor/ Moderate/ Major). If you found the set of questions in the old guidance to be confusing, you’ll be pleased that the new (2023) guidance has simplified this to Basic or Enhanced level of documentation, based on the safety risk of the software. In Section VI of the new guidance there is a table showing which software documents are required for each level. Importantly, software submissions to FDA now require the product’s full Risk Management File (plan, risk assessments, report, etc.) instead of just a “Device Hazard Analysis.” The new guidance also provides more detailed explanations of expected contents of each type of software documentation (Software Requirements Specification, Software Architecture, etc.). It also has a series of examples of medical device software and whether they fall in the Basic or Enhanced category.
Don’t forget about cybersecurity! Not mentioned directly in this guidance, but very important for anyone preparing a submission: the new FDA cybersecurity guidance (SEP 2023) requires significantly more documents in a software submission, beyond what’s called for in this main FDA guidance.
Off-The-Shelf Software Use in Medical Devices
The basic message of this guidance is that medical device companies are responsible for all of the software in their products, including software libraries and other off-the-shelf (OTS) software components that were bought instead of developed. Responsibility in this case entails defining (documenting) what OTS software you are incorporating into your product software, analyzing the safety risks associated with the OTS software, and managing changes and bugs that are discovered in the OTS software. The guidance describes the documentation to be included in submissions to the FDA as “basic documentation” for all OTS software and “special documentation” for OTS software with safety risks. The latter depends on the software Level of Concern (Major / Moderate / Minor). Cybersecurity, which was not much of a concern for medical devices when this guidance was originally published in 1999 (updated in 2019), is now an important part of risk analysis of OTS software incorporated into medical devices.
Heads Up! New FDA Software Guidances
FDA recently released these important final and draft guidances for software (as of NOV 2023):
- Content of Premarket Submissions for Device Software Functions (2023)
- Cybersecurity in Medical Devices: Quality System Considerations and Content of Premarket Submissions (2023)
- Computer Software Assurance for Production and Quality System Software (2022) – DRAFT
Each guidance shows what FDA is thinking in these areas and marks a significant change from past expectations by FDA. In other words, FDA is modernizing its expectations and recommended methods to keep up with challenges of modern software technologies and practices. Anyone developing medical device software needs to modernize as well.
Typical software documentation
So what documents does your SW team need to produce? The answer, unfortunately, is rarely a simple list. The required documents, document names, and approaches all vary depending on the company, its quality system, and its products. But they all seek to answer these essential questions:
- What are our goals for this SW development project?
- How are we organizing the project for success?
- What does the SW need to do?
- How can the SW hurt someone?
- How have we designed the SW?
- What is the test evidence that the SW does what it needs to do?
- What are the remaining bugs and are any of them a safety risk?
- What SW was tested?
- What changes were made between tests?
- How do we make sure the right SW is installed in the product?
(For example, the Software Requirements Specification document answers question #3)
Now, how much documentation is enough? How detailed do these documents need to be? These questions need to be asked for every SW development project and the answer is inherently linked to the safety risks associated with the software. A quick summary of typical software documentation is shown in this blog post: Documentation for Medical Device Software.
About the Author


Aaron Joseph helps clients to efficiently comply with regulatory requirements for medical device development. He assists clients with all aspects of design controls: risk management, requirements management, V&V testing, refining quality system procedures, and training R&D staff.
Aaron is an avid promoter of lean and agile methods for efficient product development. He helps startups and large companies to leverage requirements management tools (ALM systems) and other software tools to automate DHF documentation.
Aaron has over 20 years of experience in medical device development over a wide range of products: surgical robotics systems, laser eye surgery system, wearable device for drug adherence, digital x-ray fluoroscopy system, drug inhaler devices, x-ray catheter for brachytherapy, heart-lung bypass machine, and multiple IOT and SaMD products. This broad experience is now incorporated into Adaptive Design Controls, an integrated set of procedures to support development of complex, software-intensive medical devices.
Aaron has a BS in Electrical Engineering from Rice University and a MS in Bioengineering from University of Washington.
Pingback: Fda Validation - General Principles Of Software Validation | Fda
Aaron – great summary of IEC 62304. I appreciate the article.
Pingback: Intro to SaMD - Sunstone Pilot, Inc.