
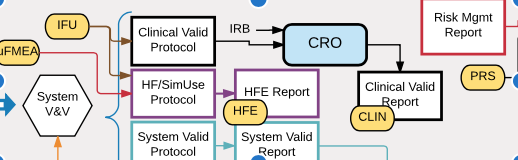
Compiling all of the DHF documentation for a new medical device can be confusing, even for experienced engineers. That’s why I created this “map” of document interdependencies. It’s a way to help explain graphically how the many documents fit together for a new product. Some key points in the map are:
- Risk analysis drives many design requirements
- V&V testing depends on the release of many documents as well as hardware and software before it can begin
- Software documentation gets special scrutiny and needs to be carefully integrated with overall product documentation
- The 510(k) submission depends on completion of a broad set of DHF documents
This Documentation Map is based on a hypothetical medical device with hardware and software components, including sterile disposable components (i.e. a Class II interventional device with embedded software). The exact set of documents needed will vary across medical devices and quality systems, but will likely follow the outlines of this map if the device includes software.
[NOTE: we are currently updating this map to include recent changes to FDA software submissions and cybersecurity documentation (map was last modified in 2017 and needs to be brought up-to-date). Stay tuned.]
| Document Title (+ acronym) | Description |
| Applicable Standards List | Defines all of the standards to which the new product must comply (e.g., ISO 10993 for biocompatibility); often combined with the PRS. |
| Clinical Validation Protocol | Defines clinical testing with representative users (i.e. clinicians) in a clinical environment; includes subject recruitment, clinical sites, IRB approval (if needed), training of subjects. |
| Component Specifications | Detailed definitions of physical characteristics of components used to support purchasing, IQC, and mfg test |
| Design FMEA (dFMEA) | Summarizes risk analysis of hardware failures and features to reduce risk; identifies safety-critical HW components; organized around product functions or in more detail around hardware components. |
| Design Review Records | Each Design Review will produce a set of files such as slides, attendance sign-in sheet, supporting data, etc. Depending on the complexity of the product, a development project may create many Design Review Packages. Some quality systems define a “Phase End Review” as a special type of design review that confirms completion of all deliverables for a defined phase of product development. |
| Engineering Reports | Reports on benchtop testing of prototypes (to guide design of commercial product) |
| Hardware Detailed Design (HDD) | Describes the mechanical and electrical design (how hardware will fulfill its requirements) |
| Hardware Requirements (HRS) | Defines product functionality allocated to hardware in more detail than Product Requirements; provides the basis for hardware design and hardware design verification. |
| Hardware Verification Protocol | Defines testing of hardware requirements; contains a series of test cases, as well as test environment, test tools, description of test articles, and other information for execution of the test cases; testing can be performed on full product or a subassembly. |
| Human Factors (HF) / SimUse Protocol | Defines usability testing with representative users (i.e. clinicians); includes subject recruitment, usability labs, IRB approval (if needed), training of subjects. |
| Human Factors Evaluation (HFE) Report | Summarizes the identification, evaluation, and final assessment of all serious use-related hazards for the product. |
| Human Factors Formative reports | Reports on early evaluation of prototypes by representative users (to guide design of user interface) |
| Label Specifications | Detailed design of the labels on the product and product packaging, including artwork needed for printing |
| Labeling Requirements (LRS) | Defines content of product and packaging labels and user manual (warnings, symbols, etc.) |
| Master V&V Plan | Covers all V&V testing–roles, responsibilities, activities, deliverables; entrance and exit criteria; may have multiple test plans underneath it. |
| Process FMEA (pFMEA) | Summarizes risk analysis of manufacturing process failures and the measures to reduce risk; organized around manufacturing workflow. |
| Product Bill of Materials (BOM) | A structured list of all of the components, labels, etc. for the product which is used as a basis for manufacturing. |
| Product Development Plan (PDP) | Describes at a high-level everything needed to develop the new product; includes roles, responsibilities, activities, and deliverables; interfaces with development partners and other service providers; updated throughout development as needed. |
| Product Requirements (PRS) | Top level design requirements for the new product (includes Intended Use and User Needs); provides a user’s view of device (instead of an engineer’s view). |
| Requirement Trace Matrix | Shows that all requirements were tested |
| Risk Management Plan | Describes all activities for product development focused on analyzing and reducing safety risks per ISO 14971. |
| Risk Management Report | Summarizes all risk management activity during development of the new product and remaining risks associated with it; defines all documents that constitute the Risk Management File for the product (per ISO 14971); defines or references procedures for risk management after product launch. |
| Risk Trace Matrix | Shows that all risk controls were implemented and tested |
| Software Detailed Design (SDD) | Describes the design of the software including state diagrams, data structures, interfaces, etc (how software will fulfill its requirements) |
| Software Development Plan (SDP) | Describes software activities, responsibilities, and deliverables for development of the software for the new product; defines details of software design control procedures; can be combined into the PDP. |
| Software FMEA (sFMEA) | Summarizes risk analysis of software failures and features to reduce risk; organized around software functions or software components. |
| Software Release Package | Set of records released with the software which includes summary report of SW testing, SW revision history, and list of open SW bugs. |
| Software Requirement Trace Matrix | Shows that all software requirements were implemented and tested |
| Software Requirements (SRS) | Defines product functionality allocated to software (more detailed than Product Requirements); provides the basis for software system verification testing. |
| Software System Verification Protocol | Defines testing of software requirements; contains a series of test cases, as well as test environment, test tools, and other information for execution of the test cases; includes software integration testing. |
| Software Unit Verification Summary | Report summarizing detailed verification of software components, including code reviews, static analyses, and unit testing. |
| System Architectural Design (SAD) | A high level description of the product design; shows a breakdown into modules/components with key interfaces; includes definition of the SW Level of Concern (per FDA) and the SW Safety Classification (per IEC 62304). Products with complex software may have a separate SW Architectural Design document (SWAD). |
| System Hazard Analysis (SHA) | Top-down, comprehensive analysis of risks associated with the product; defines necessary risk controls (safety features) to reduce risk; these are traced to design requirements (product/ software/ hardware). |
| System Verification Protocol | Defines testing of full product against product requirements (testing typically performed by test engineers). |
| Test Equipment Validations | Reports demonstrating that a particular tool, meter, or fixture used in V&V testing is sufficiently accurate and reproducible; includes SW test tools/test scripts. |
| Test Method Validations | Reports demonstrating that a method/technique of measurement is sufficiently accurate and reproducible (for example measurement of fluid flow from a pump) |
| Use FMEA (uFMEA) | Summarizes risk analysis of use error and features to reduce risk; organized around user workflows. |
| User Manual / Instructions For Use (IFU) | User documentation that explains how to use the product, as well as critical warnings and cautions associated with safety risks; can be in printed or electronic form (or a combination of the two). |
Originally posted at http://consensiainc.com/2017/04/27/zipquality-medical-device-documentation-map/
Pingback: Newsletter V. 2019 Issue 1 - Sunstone Pilot, Inc.
Pingback: Documentation for Medical Device Software - Sunstone Pilot, Inc.