

December 2021 Newsletter – Digital Health – Big Updates from FDA

Big news! After 16 years, the FDA is changing its guidance on the software documentation needed in regulatory submissions. The new draft guidance, “Content of Premarket Submissions for Device Software Functions,” was announced last month. When finalized, this new guidance will supersede the Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices (May 2005).
This new draft guidance is quite different from the 2005 guidance. Here are some key changes in it:
This new draft guidance is quite different from the 2005 guidance. Here are some key changes in it:
- The documentation will no longer be determined by software Level of Concern (Minor / Moderate / Major); instead, it will be determined as needing Basic or Enhanced level of documentation (based on the software’s risk). The Enhanced level is required if any of the following apply:
- Class III or combination device
- Device tests blood donations or determines donor-recipient blood compatibility.
- Device software functions could present a probable risk of death or serious injury (pre-mitigations)
- The Device Hazard Analysis is replaced by the device’s Risk Management File documents.
- The new guidance allows for the declaration of conformity to the IEC 62304 medical software standard.
- The new guidance has more explanations of what is required in each type of software document, such as the architecture diagram.
For more information on the changes, I recommend this summary by John Murray of SoftwareCPR: New FDA draft Software Premarket Submission Guidance
The Software Landscape
At a medical device company, there can be many types of software, and it’s often confusing to know what are the regulatory requirements for each type and what documentation is needed. The diagram below summarizes the different types of software with some examples of each. The types are split broadly into Product vs. Non-Product software because the regulations and standards are split that way (e.g., the IEC 62304 standard only applies to Product software). The regulated Product software is divided by safety classification (A/B/C).
For Non-Product software, it can be difficult to know what is regulated and what is not. The key consideration here is determining if the software automates or otherwise supports a regulated process. For example, software that manages document approvals and changes for Doc Control is regulated and needs to be validated for its intended use.
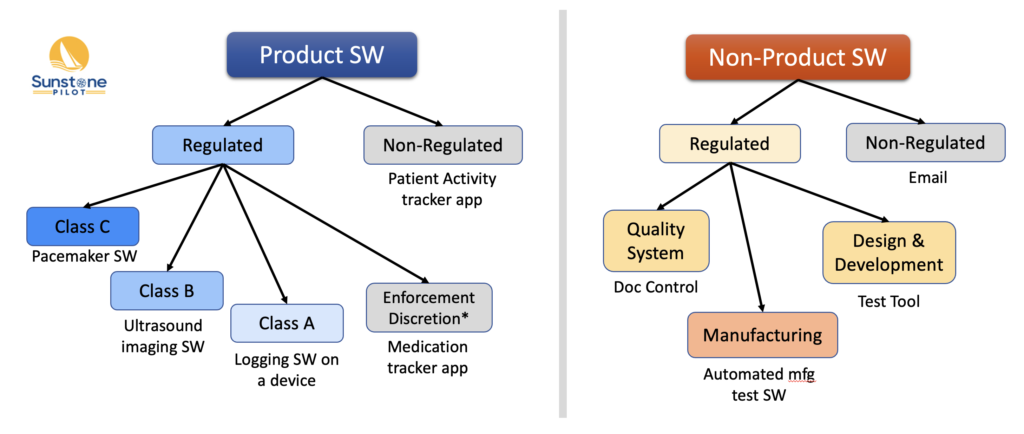
*Enforcement discretion means that FDA regulations apply but the FDA has decided not to enforce them for certain low risk software.
Confusingly, many modern medical devices, especially in digital health, include both regulated and non-regulated product software. For guidance on managing a mix of regulated and non-regulated software functions, see the FDA guidance Multiple Function Device Products: Policy and Considerations (JUL 2020).

Playbook for Threat Modeling for Medical Devices – a downloadable resource for developing or evolving a threat modeling practice throughout your organization. Contains detailed explanations of multiple methods for cybersecurity analysis and protection, based on a series of workshops sponsored by the FDA.
Sunstone Pilot™ On-demand Training
Sunstone Pilot offers flexible, cost-effective training for medical device design controls, allowing you to train as many or as few employees as you need—from a single team or department to everyone in your organization.
We offer both standardized training as well as training customized to the specific needs of your organization.
Upcoming Events
BIOMEDigital
December 8-9 2021, San Jose, California
https://www.biomedevicesanjose.com/en/home.html
IMPACT Boot Camp: Navigating the Journey from Digital Health Technologies to Meaningful Patient Outcomes
CDRH, Johns Hopkins University CERSI, and the University of Maryland CERSI offer an immersive mini-bootcamp
January 26, 2022
Bay Area Biomedical Device Conference
March 29-30, 2022
Hosted by SJSU in San Jose, California