

Introduction
How do we manage upgrades to a new medical device after product launch? If you don’t expect to ever make changes to the product then this may not be a concern at all. However, if you’re developing software-intensive, connected devices or capital equipment then this is a big concern. The majority of development projects will likely be product upgrades after initial launch and your quality system should be designed with this in mind.
What is meant by “re-entrant design controls”? It means designing quality system procedures around the expectation of regular product upgrades and repeating design control phases. It helps to understand this by starting with a description of a typical framework for medical device design controls. Design controls for medical device development are typically organized around a series of phases (Phase 1, Phase 2, etc.) with gates between them such as in the diagram below. The phases may vary in name and number from company to company but the basic idea is the same. Each phase has specific requirements that a product team must fulfill to complete the phase. When the team completes all the pre-market phases they can release the product to market. And along the way they will generate all the documentation needed for the product’s Design History File (DHF).
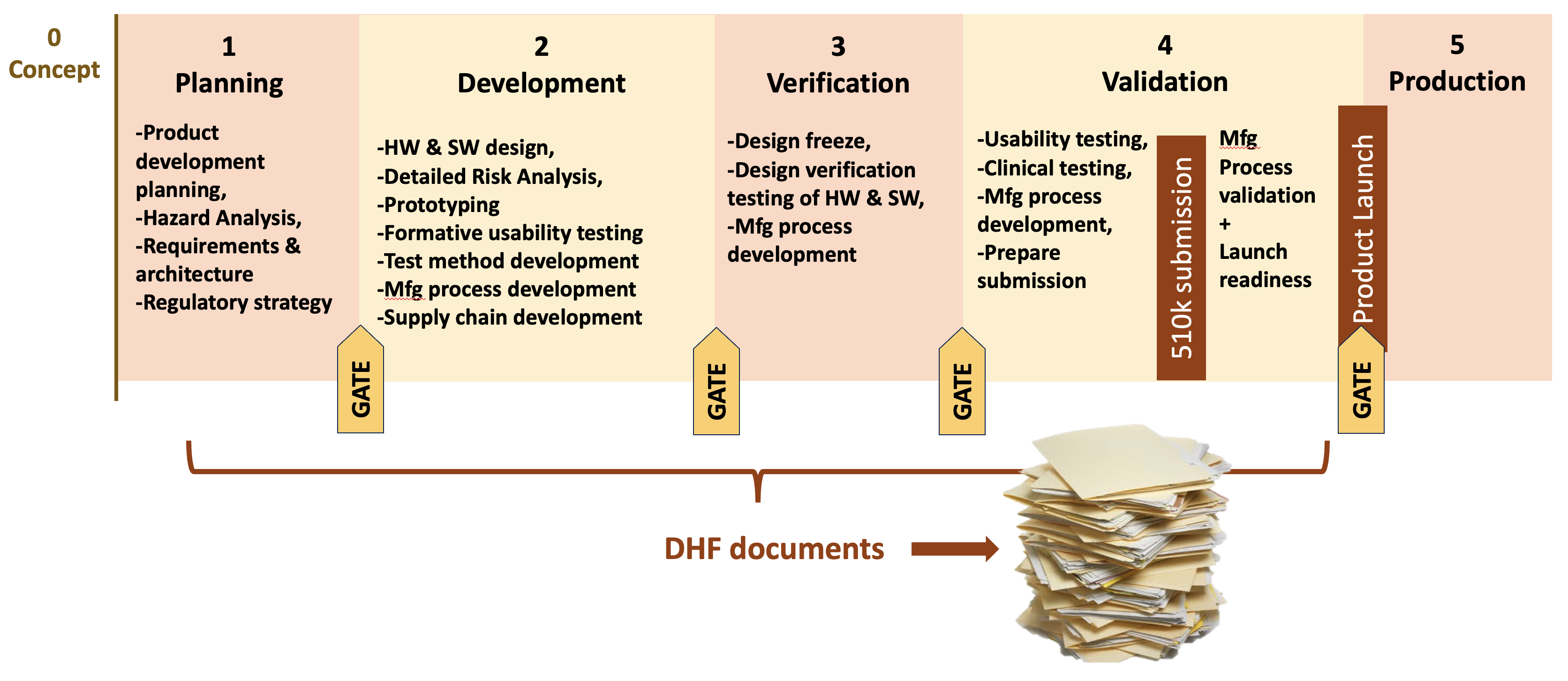
As a consultant, I see many quality systems and different approaches to design controls. I regularly come across quality systems that assume a product will be developed once and essentially never upgraded (what I refer to as a “once-though” approach). The procedures include simple methods for managing small post-market changes to the product (e.g. change to a supplier) but nothing that would apply design controls systematically to more extensive changes. This doesn’t mean that the “once-through” approach is wrong. It will work fine for a surgical instrument or sterile disposable. But it doesn’t work for many software-intensive, connected medical devices or for capital equipment.
For example, consider a hypothetical company manufacturing endoscopes for laparoscopic surgery. Let’s say the company has just launched a new endoscope system. The product works fine but after launch one of the engineers identifies a better illumination source that can provide 30% brighter light with a wide range of tints (color temperature). The head of Marketing pushes to get the product upgraded with this new illumination source right away because it is a clear performance improvement and the manufacturing changes are simple. Can the change to the illumination source really be treated as just swapping out one module for another? What is the full impact of this change to the product and the users? Should the user interface change? What kind of testing will be needed? Which existing DHF documents need to be updated and what new documents are needed? Could this trigger a new regulatory submission?
Alternatively, let’s say the endoscope system exports video and images to a hospital PACS system. When the PACS software is updated, with a slightly different data interface, the endoscope system software will need to be updated as well to continue communicating properly. How should this product change be managed?
These questions are not simple to answer and product upgrades are not simple to manage. They require more than a change impact form or ECO to manage the upgrades. To ensure a safe and effective medical device, the team often needs to start back in Phase 1 and repeat some or all of the design controls. This is what I call “re-entrant design controls” and it means designing quality system procedures around the expectation of regular product upgrades and repeating design control phases.

Principles
Fully supporting re-entrant design controls requires attention to multiple aspects of the quality system. Here are 4 principles to incorporate in your quality system to support re-entrant design controls, with the most important principle being predetermined change pathways.
Predetermined change pathways
>Many companies rely on a one-size-fits-all approach which over-manages small changes and under-manages large changes (and often involves lots of meetings and arguments for each change project). With pre-defined pathways every proposed post-market change can be efficiently managed and each pathway can be optimized over time (e.g., with checklists).
The number of pathways to define depends on a company’s products and organization. For example, one company may find that 3 pathways are optimal; other companies may choose 4 or 5 pathways. For our hypothetical endoscope manufacturer, let’s assume they have 4 pathways. Here they are described as T-shirt sizes:
- X-Small: Software patch / hotfix – expedited software release to correct a software bug or a cybersecurity vulnerability
- Small: Hardware component changes
- Medium: Minor upgrade – limited hardware and/or software changes; a subset of design controls apply
- Large: Major upgrade – most design controls apply (and likely needs a re-submission before release)
With pre-defined pathways the complex questions of how to manage a product upgrade boil down to just: “Does the proposed change fit the criteria for Pathway X?” For example, we can now determine that the change to the endoscope illumination source will be a “Major upgrade” that will be managed with a new plan and will involve most of the design controls and will repeat all phases. In contrast, the software change to maintain operability with the external PACS system could be handled as a quick software patch. Note that it’s important to clearly define the boundaries/ requirements for each pathway. Otherwise, project managers will be tempted to choose the fastest pathway for any upgrades rather than the most appropriate pathway.
Flexibility through development plans
The Standard Operating Procedures (SOPs) in the quality system provide the framework for development projects and should state the full requirements for new product development. However, the required activities and deliverables for a product upgrade will be different than for new product development. Some existing DHF documents may need to be updated (Risk Management File, Product Requirements, etc.) and new documents created (test reports, software release records, etc.). Therefore, the quality system needs a way to modify the requirements for each project. The Product Development Plan (AKA “Design and Development Plan” or “Project Plan”) is the mechanism to do this. The SOPs define the default requirements for all development projects and then each plan determines the activities and deliverables for a particular project within the framework. The plan can scale up or down the design controls for bigger or smaller projects, providing inherent flexibility in the quality system procedures where needed. This is much more effective than what I would call “negotiated flexibility” where the team needs to get explicit permission for every project to deviate from detailed requirements in an SOP (What happens if the head of Quality is replaced in the middle of the project after negotiating with his or her predecessor?).
DHF organized for long-term record retrieval
The way your quality system organizes Design History Files (DHFs) will impact how well it supports re-entrant design controls. Many DHFs are organized around phases: a set of all the documents for Phase 1, a set for Phase 2, etc. This is problematic if projects repeat phases and generate many new documents over years of upgrades. For example, how can we know if the documents in the Phase 2 set apply to the originally released product or to one of the (many) product upgrades?
Instead, the DHF documents should be organized by category, not by phase. For example, a set of design planning documents, a set of design requirement documents, a set of software verification documents, etc. With DHFs organized this way, after many product upgrades over many years we can still navigate the DHF and identify which DHF documentation applies to which version of the product.
Efficient document release and revision
Being able to manage many changes to a product after launch means being efficient at document revisions and release. If it takes 10 days or more to approve and release documents at your company then you will find it very difficult to, for example, perform expedited software patches (to address a cybersecurity vulnerability, for example). Revising procedures to minimize delays in review, approval, and release can have a huge cumulative effect in efficiently managing product changes. Do we really need 8-10 signatures on every document? And while you’re revising procedures, look for redundant records that can be streamlined. Nobody wants to enter the same information in multiple documents.
Conclusions
There are many ways to implement medical device design controls and still comply with applicable regulations and standards. Some procedures define a once-through approach (design control phases never repeat) and some the re-entrant approach. For software-intensive, connected medical devices or capital equipment, re-entrant design controls will provide a more systematic and efficient way to manage the many expected changes throughout a product’s lifetime. Product teams benefit from clear procedures that scale to manage product upgrades based on the nature of the changes. Establishing those procedures is straightforward once the company embraces the concept of expecting and supporting regular product upgrades.
Want re-entrant design controls but don’t want to spend months revising SOPs? The Adaptive Design Controls package incorporates re-entrant design controls, support for agile software development, and adapts to the software tools your product teams use.