
Last modified: 12-NOV-2025
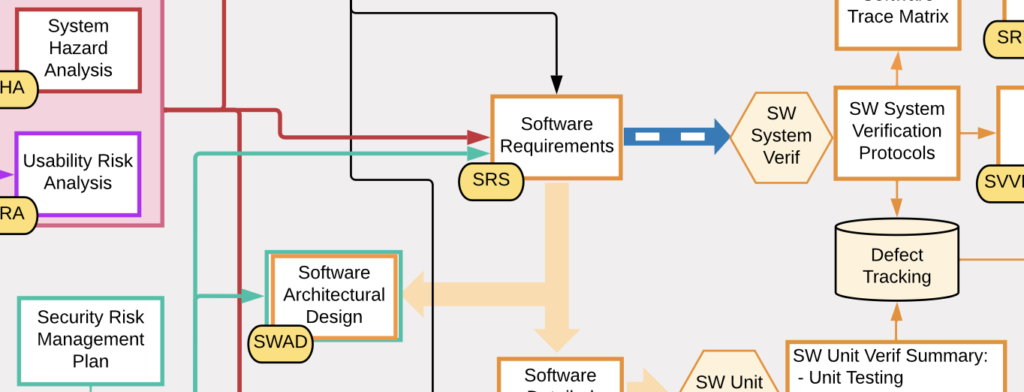
Software as a Medical Device (SaMD) products do not involve hardware or manufacturing but their documentation is still complicated and it can be difficult to know what is truly required for compliance. To help reduce the confusion, I created this SaMD Documentation Map which illustrates the formal documentation needed for a typical SaMD product. The map is a graphical representation of the interdependencies of the product documentation.
The map reads roughly chronologically from left to right (yes, I know that software development is iterative but for simplicity no iterations are shown in the map). Each arrow represents a dependency between documents (i.e. the document the arrow points to cannot be completed without the information in the document the arrow originated from). The items on the far right are sections of an FDA submission.
Some key points in the map are:
- Risk analysis and cybersecurity drive many design requirements
- V&V testing depends on the release of many documents as well as completed software before it can begin
- There are key documents at the product/system level in addition to all of the software-specific documentation (colored orange)
- An FDA 510(k) submission (and other submissions) depend on completion of a broad set of DHF documents

This Documentation Map is based on a hypothetical software-only medical device. The exact set of documents needed will vary across SaMD products and quality systems, but will likely follow the outlines of this map, even if some of the document names change. The table below describes each document and other items in the map.
| Document_Title_(acronym) | Description |
|---|---|
| Clinical Validation Protocol | Defines clinical testing with representative users (i.e. clinicians) in a clinical environment; includes subject recruitment, clinical sites, IRB approval (if needed), training of subjects. |
| CRO | Contract Research Organization: An organization that offers clinical trial and related services for medical device or pharmaceutical testing |
| Cybersecurity Risk Analysis (CRA) | A security risk assessment of the product being developed based on the threat modeling; includes analysis of system assets and their vulnerabilities, risk scoring, and identification of risk controls |
| Cybersecurity Architecture Views | Defines the system and its interfaces from a cybersecurity viewpoint, with high-level definitions of the system assets and interactions with other devices and/or systems; design information on how those interactions occur and are secured; secure software patches/updates |
| Cybersecurity Post-Market Management Plan | The post-market plan for maintaining device security, including cybersecurity monitoring, metrics, labeling, controls, incident response, software patches and updates, and coordinated vulnerability disclosure |
| Design Review Records | Each Design Review will produce a set of files such as slides, attendance sign-in sheet, supporting data, etc. Depending on the complexity of the product, a development project may create many Design Review Packages. Some quality systems define a “Phase End Review” as a special type of design review that confirms completion of all deliverables for a defined phase of product development. |
| Engineering Reports | Reports on early testing of prototypes (to guide design of commercial product) |
| Human Factors Evaluation (HFE) Report | Summarizes the identification, evaluation, and final assessment of all serious use-related hazards for the product. |
| Human Factors / Usability Formative reports | Reports on early evaluation of prototypes by representative users during development (to guide design of user interface) |
| Human Factors / Usability Summative Protocol | Defines usability testing with representative users (i.e. clinicians); includes subject recruitment, usability labs, IRB approval (if needed), training of subjects. |
| Human Factors / Usability Summative Report | Final report on usability testing, as part of system validation, confirming that the design of the product’s user interfaces meets user needs |
| Instructions For Use (IFU) | (see User Manual) |
| IRB | Institutional Review Board: a group that has been formally designated to review and monitor biomedical research involving human subjects; reviews and approves clinical plans/protocols before beginning a clinical trial |
| Labeling Requirements | Defines content of product and packaging labels and user manual (warnings, symbols, etc.) |
| Product Development Plan (PDP) | Describes at a high-level everything needed to develop the new product; includes roles, responsibilities, activities, and deliverables; interfaces with development partners and other service providers; updated throughout development as needed. |
| Risk Management Plan (RMP) | Describes all activities for product development focused on analyzing and reducing safety risks per ISO 14971. |
| Risk Management Report | Summarizes all risk management activity during development of the new product and remaining risks associated with it (per ISO 14971); defines or references procedures for risk management after product launch. |
| Security Assessment of Unresolved Anomalies | An analysis of the open bug list from a security viewpoint; shown combined with the Software Unresolved Anomalies Report (with safety risk assessments) |
| Security Risk Management Plan | A description of the security activities and deliverables during product development, including roles, responsibilities, and resources; defines how threat modeling and cybersecurity risk assessment will be performed. This plan is separate from and complementary to a safety Risk Management Plan for ISO 14971. |
| Security Risk Management Report | The report summarizes the risk evaluation methods and processes, describes the risk mitigation activities and residual security risk, describes the results of security testing, and provides traceability between the threat model, cybersecurity risk assessment, SBOM, and testing documentation. |
| Software Bill of Materials (SBOM) | A formal inventory of software components and dependencies, information about those components, and their hierarchical relationships; the basis for vulnerability scanning |
| Software Detailed Design (SDD) | Describes the design of the software including state diagrams, data structures, interfaces, etc. (how the software will fulfill the software requirements); also referred to as Software Design Specification |
| Software Development Plan (SDP) | Describes software activities, responsibilities, and deliverables for development of the software for the new product; defines details of software design control procedures; can be combined into the PDP |
| Software Release Record (SRR) | Identifies the software being released; includes software release notes, summary of SW testing, SW version history, and list of open SW bugs |
| Software Requirements Specification (SRS) | Defines product software functionality (more detailed than User Requirements); provides the basis for software system verification testing |
| Software System Verification Protocol | Defines testing of software requirements; contains a series of test cases, as well as descriptions of the test environment, test tools, and other information for execution of the test cases; includes software integration testing; often multiple protocols are written to cover testing all of the requirements |
| Software Trace Matrix | Table linking software requirements to software tests and software design; shows that all software requirements were implemented and tested. |
| Threat Model | A structured process which identifies, analyzes, and prioritizes potential threats and vulnerabilities in a system |
| Usability Risk Analysis (URA) | Risk assessment of use error and features to reduce risk; organized around use scenarios and user tasks; drives summative usability testing; common alternative names are Use FMEA and Use Related Risk Analysis (URRA) |
| User Manual / Instructions For Use (IFU) | User documentation that explains how to use the product, as well as critical warnings and cautions associated with safety risks; can be in printed or electronic form (or a combination of the two). |
| User Requirements Specification (URS) | Top level design requirements for the new product (includes Intended Use and User Needs); provides a user’s view of the device (instead of an engineer’s view). |
Sign Up for Updates
Sunstone Pilot’s Best Practices Newsletter
Revision History
| Version of Map | Date | Summary of Changes |
|---|---|---|
| v1.3 | 12-NOV-2025 | Separated Cybersecurity Architecture Views (CAV) document from Software Architectural Design (SWAD) to clarify their complementary roles; renamed Software Security and Maintenance Plan to Cybersecurity Post-Market Management Plan to avoid confusion with other plans; added Software Test Plan; added Vulnerability Scan of SBOM; revised list of prerequisites for starting V&V |
| v1.2 | 28-JAN-2025 | Showed SBOM dependency on software design |
| v1.1 | 02-DEC-2024 | Multiple corrections based on reader feedback; expanded legend; revised naming of some FDA eSTAR sections |