

Beginner’s Guide to Medical Device Software
“Medical Device Software for Newbies” or “Why is this stuff so confusing?”
Many times I’m asked to explain to engineers how to manage compliance for medical device software development. From newcomers to experienced professionals, it remains a source of confusion and frustration. And a lot of the confusion comes from the special terminology, particular regulations and standards, and ambiguous or conflicting quality system procedures. Medical device software has its own words, rules and methods. The goal of this blog, then, is to remove the mystery and myth from managing medical device software.
This blog is designed for people who are new to medical device software—either experienced with software but not medical device regulations or experienced with medical device hardware only.
Software is Special!
If software is involved in your product, expect special requirements for compliance. The FDA states that “because of its complexity, the development process for software should be even more tightly controlled than for hardware, in order to prevent problems that cannot be easily detected later in the development process” (General Principles of Software Validation guidance). In contrast to hardware changes, with software, you can easily make significant changes to your product that go unseen to the naked eye. The FDA, in response to this issue, has declared that “… software engineering needs an even greater level of managerial scrutiny and control than does hardware engineering.”
What is it?
So what is this mysterious entity, anyway? In a nutshell, “medical device software” is the software in your product. It could be software that acts as an accessory to a medical device or software that is itself the medical device (without hardware). It’s a straightforward definition that quickly becomes complicated when talking about networked devices and other forms of software like mobile apps (but that’s for another blog post).
Another way of defining medical device software is by explaining what it is not. It is not software used in manufacturing, such as automated test equipment, nor is it software used in the quality system, such as managing CAPAs, doc control, training records, etc. These other categories of software used in a medical device company, frequently referred to as “non-product software”, are still regulated but not by the same regulations as medical device software.
What are the rules?
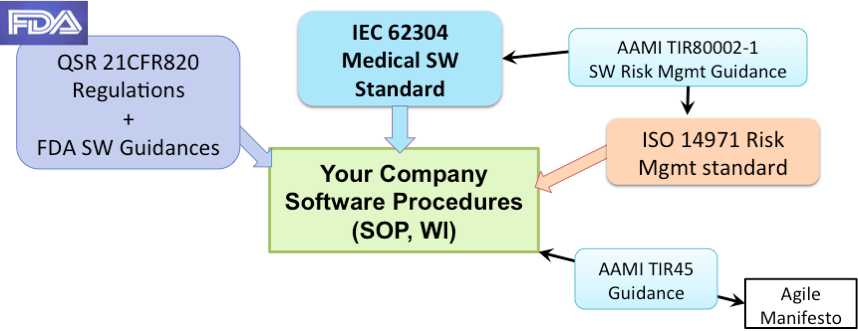
This is one of the big sources of confusion with medical device software: there is no single set of rules. Software, as I mentioned before, has special requirements and is given a greater level of scrutiny, but, the general requirements are not new. Instead of writing additional, new regulations for software, the FDA issued multiple guidances over the past 20 years on how to apply existing regulations (21 CFR 820) to multiple types of software. Additionally, and confusingly, there exists the IEC 62304 Medical Device Software standard. Both sets of rules overlap but use different terminology. My recommendation is to base your software development procedures on the IEC 62304 Standard, which is easier to understand, and then include any additional adjustments needed to meet all the FDA requirements.
The diagram below summarizes these key regulations and standards for medical device software development. Note that your software development procedures must include appropriate risk management. The AAMI TIR80002-1 provides clear guidance on how to apply risk management to medical device software. The diagram also includes the AAMI TIR45:2012 guidance which is important if you want to incorporate agile methods into your medical device software development.
What is Software Validation ?
This is where things get really confusing. Both design verification and design validation can have different meanings when applied to software instead of hardware. When we test hardware against requirements (i.e. bench tests of a mechanical assembly) that’s referred to as “design verification” not validation. However, many medical device companies define testing software against software requirements as “software validation.” The FDA guidances have not made this any clearer since their definition of software validation has evolved over the years.
I recommend using definitions similar to the ones below since they are more consistent with general definitions of design verification and design validation and they make it easier to understand the IEC 62304 standard:
- Software verification = confirmation through provision of objective evidence that specified requirements have been fulfilled
- Software validation = confirmation through provision of objective evidence that software specifications conform to user needs and intended uses, and that the particular requirements implemented through software can be consistently fulfilled
Importantly, the IEC 62304 standard only covers software verification (per the above definitions). Software validation therefore becomes part of the validation testing of the entire product, such as usability testing and clinical evaluation, instead of being performed as a separate software-only activity.
Within the broad category of software verification, IEC 62304 calls out three main levels of SW testing:
- SW unit verification – lowest level testing; frequently performed by the developer on a portion of the code in a desktop test environment; unit verification also includes activities such as code reviews and static analysis
- SW integration testing – combining software units/modules and combining software and hardware (running on actual product hardware); frequently performed by a combination of developers and SQA
- SW system testing – testing software against software requirements; usually performed by SQA to formal (released) test protocols
Keeping in mind these 3 levels of software testing will make it easier to plan software development and determine which activities are solely the responsibility of the software group and which are shared with the rest of the product development team.
One more important point: at a medical device company whose products have no software, the quality system procedures for “software validation” will likely only be applicable to non-product software (also referred to as “computer system validation”).
In the next blog post we’ll dive into some details of the IEC 62304 software standard and FDA software guidances to better explain some of the issues discussed here, but hopefully this can act as a good starting point for navigating through the complicated realm of medical device software.
I agree with Aaron that 62304 and the FDA Guidance sometimes seem at odds, or at least cause confusion by omission. With respect to software validation, 62304 is pretty clear that that standard does not address validation of the final device, even when the device consists entirely of software (cf. 62304, section 1.2). Elsewhere in 62304, the standard states that software is a subsystem of a device and validation refers to testing the whole device against device requirements; this is especially clear in Annex C in the comparison to PEMS as defined in 60601 (cf. 62304, Table C.3, 14.3). Unfortunately, in Figure C.2, which is taken from another standard, there is a box labeled “PEMS validation”, which is misleading in the context of 62304.
One other area where it is useful to stick to the definitions of 62304 are the terms software unit / item / system. The lingua franca of software development is 62304, including with the FDA, so developers should understand the definitions of these terms and use them accurately in all documents.
As Aaron said, “Software is Special.” The expectations for medical devices are higher than for consumer electronics, on par with aerospace and some aspects of automotive. Because software is less deterministic than even a zillion hardware gates it takes more planning, design, and verification to assure acceptable risk.
Pingback: Where to Start When Developing Medical Devices | Voler Systems
Pingback: Get Started: Developing Medical Devices | Voler Systems